Artificial intelligence (AI) is changing this game, shifting R&D away from resource-intensive brute-force screening toward informed navigation. Instead of designing experiments manually, scientists can now rely on easy-to-use AI-based tools that guide the search through this immense design space. These tools recommend the most informative next experiment based on results collected so far. Scientists then run the recommended experiment, feed the results back into the system, and repeat the loop until convergence.
By automating the heavy lifting of experimental design, these tools free scientists to focus on what matters most: interpretation, analysis, and creative problem-solving. The result is a more efficient, digitalized discovery process that turns every experimental outcome into compounding knowledge.
Table of Contents
- Main Takeaway: AI as an Actionable Decision Engine
- From Edisonian Trial-and-Error to the Engine of Efficiency: Bayesian Optimization
- Redefining Reaction Generalization via Transfer Learning
- Optimizing Across Information Frontiers: Multi-Fidelity Bayesian Optimization
- Physical AI in the Lab: Self-Driving Labs®
- Transforming R&D from Linear to Exponential
Main Takeaway: AI as an Actionable Decision Engine
From Edisonian Trial-and-Error to the Engine of Efficiency: Bayesian Optimization
To break through these bottlenecks, AI-assisted discovery reframes the challenge as an optimization problem. Rather than searching exhaustively, researchers first define a rigorous objective function that quantifies the desired properties (e.g., maximizing yield and selectivity while minimizing raw material costs). The system then searches intelligently across the multi-dimensional design space to optimize these objectives. Bayesian Optimization (BO) provides a principled framework for this search, using a probabilistic model of the objective to decide which experiments to run next based on what is known versus what is merely guessed.
The Mechanics Behind the Intelligent Search & Bayesian Optimization
To achieve this extreme data efficiency, the algorithm relies on a clear division of labor between two core components: a predictive model and a decision rule.
- The Probabilistic Surrogate Model (The Predictive Model): Typically driven by a Gaussian Process (GP), this functions as a dynamic digital twin of the multi-dimensional reaction landscape. Based on sparse initial runs, it estimates how the reaction will behave across untried conditions. Crucially, because it is probabilistic, it doesn’t just give a blind prediction; it quantifies its own uncertainty, mapping out where it has high confidence based on hard data and where it is merely guessing.
- The Acquisition Function (The Decision Rule): This component acts as the decision rule that scans the landscape to select the next experiment. Instead of a scientist manually weighing competing variables, the algorithm balances two core optimization criteria simultaneously:
- Exploration: Directing the next run into highly uncertain, unmapped territory to see if a superior catalyst backbone, solvent system, or formulation is hiding there.
- Exploitation: Steering toward known, high-performing regions to make small adjustments to the optimization variables and hit the peak performance target.
Other AI approaches typically lack this fit. For example, point-prediction ML models output a single best guess with no sense of their own reliability, so they need large datasets to become trustworthy and fail silently when data is scarce. For large language models (LLMs), they are trained on text in which real-world experimental data is barely represented, leaving them without grounding in the physical and chemical behavior of actual systems.
Case Study: Cutting Catalyst Costs Without Sacrificing Performance
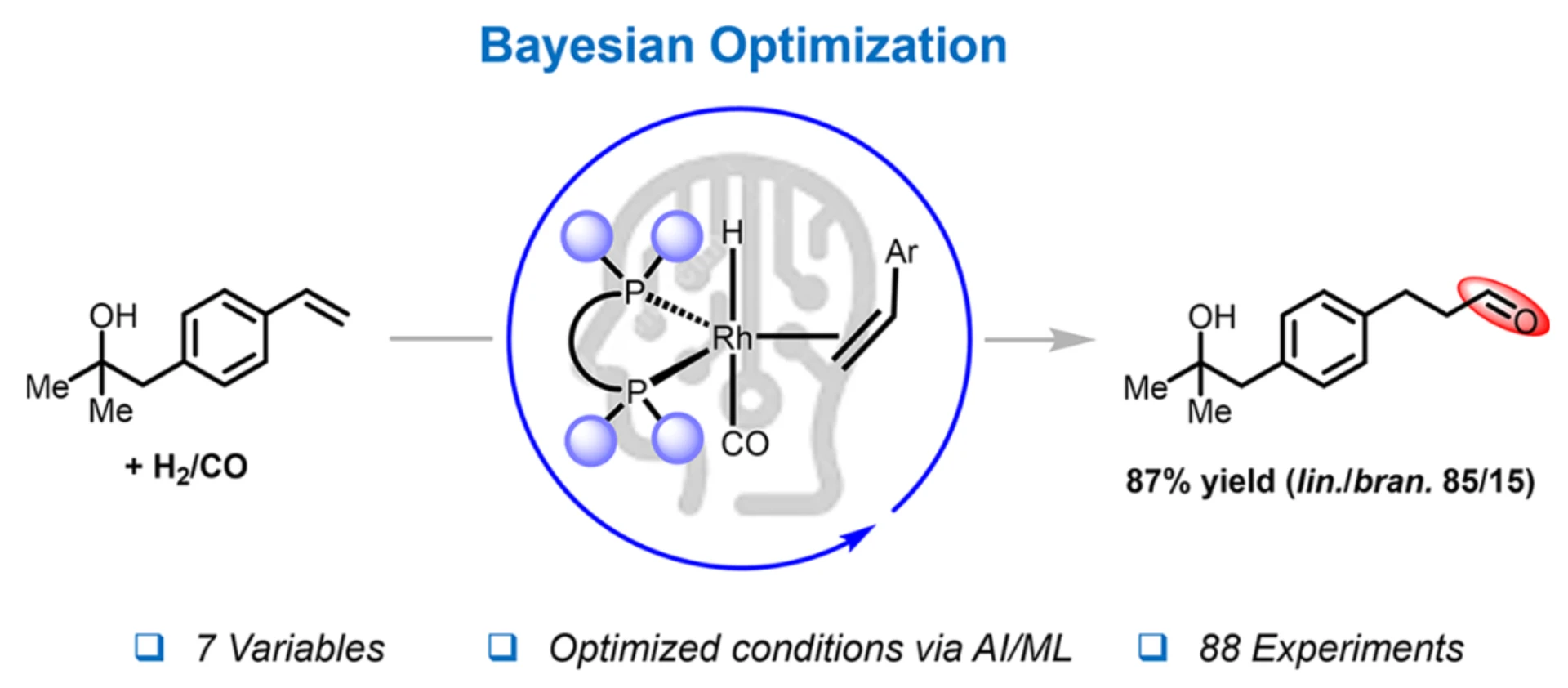
Figure from graphical abstract Saudan et al. Bayesian Optimization for Resource-Efficient Hydroformylation. ACS Catalysis. 2025.
Learning From Every Campaign:
Transfer Learning Across Substrates
Transfer learning breaks this cycle. Rather than forcing one rigid recipe onto every substrate, it maps the underlying features of the reaction landscape and carries them forward. Knowledge from past (“source”) campaigns narrows the search space for each new (“target”) campaign, so the model can converge on the right conditions for an unfamiliar substrate with only a few experiments.
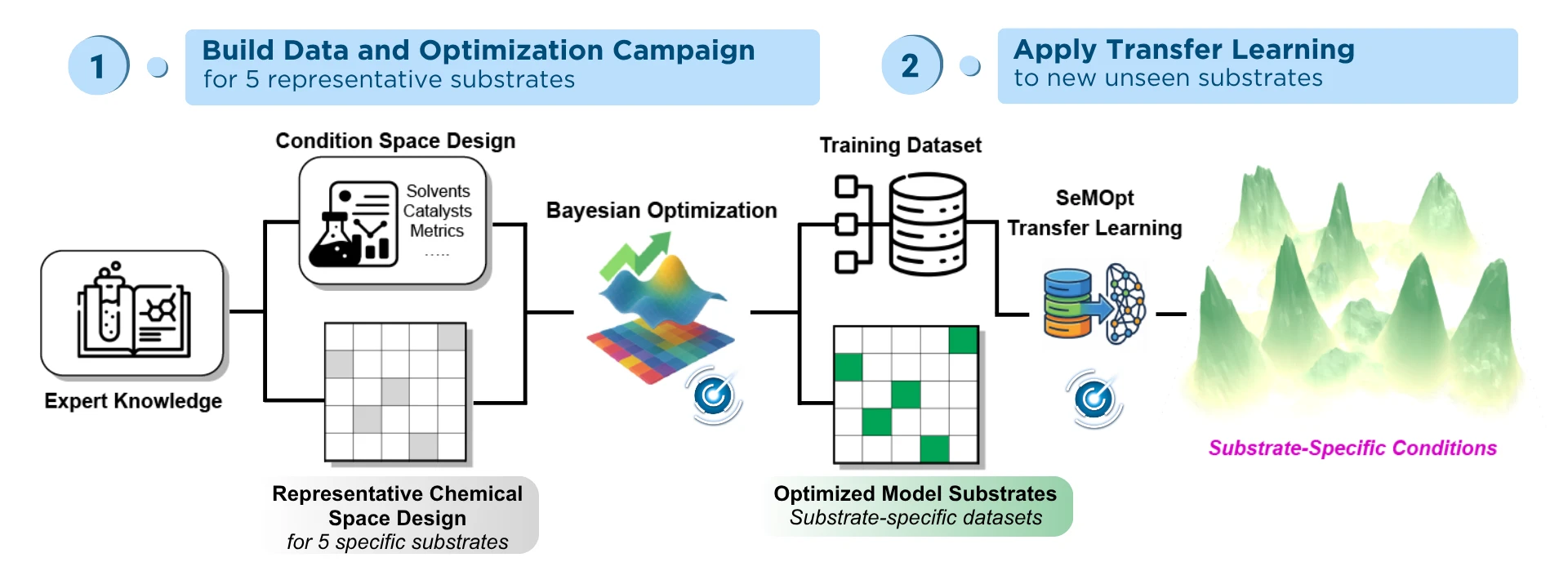
Caption: Figure adapted from graphical abstract Peng et al. Accelerating Reaction Generalization through Domain-Specific Transfer Learning. Chemrxiv. 2026.
A recent joint ChemRxiv publication between the lab of Dr. Alan Healy at NYU Abu Dhabi and Atinary. Using Atinary’s SDLabs, the collaboration provided a definitive demonstration of how transfer-learning-guided workflows can accelerate substrate-specific reaction optimization.
Instead of relying on large, literature-derived datasets that carry their own biases, the workflow operates via an efficient three-step framework:
- Explore: The team navigated the initial reaction landscape using BO in SDLabs, building a compact, domain-specific dataset of just 120 experiments from an expert-guided selection of the reaction space.
- Adapt: Using Atinary’s proprietary transfer learning algorithm, SeMOpt (Semantic Memory Optimization), this curated data was applied as “semantic prior knowledge” to entirely new, unseen substrates.
- Optimize: By balancing this semantic knowledge with targeted exploration-exploitation, the model converged on optimal, substrate-specific conditions much faster, in four experiments or fewer.
Physical AI in the Lab: Self-Driving Labs®
A definitive look at this architecture in practice is our state-of-the-art Atinary Lab in Boston. Operating as a Scientific Discovery Factory, the facility features two autonomous platforms that continuously Design, Make, Test, Analyze and Learn (DMTA+L) from real-world experiments around the clock.
Atinary’s Self-Driving Labs® integrate our cloud-native, code-free AI platform (SDLabs) with world-class automation and instrumentation from an ecosystem of industry-leading partners:
- Robotics & Automation: ABB Robotics and Chemspeed Technologies provide configurable automation systems that handle the physical preparation and execution of complex reactions with high precision.
- Precision Measurement & Synthesis: Mettler-Toledo delivers the laboratory instrumentation required for high-fidelity physical operations and sample handling.
- Advanced Analytics: Agilent (Open Bed Sample Fraction Collector and Liquid Chromatography and Mass Spectrometer) and Bruker (benchtop NMR) provide high-throughput tools that characterize reaction outputs in real time.
- Cloud Infrastructure: Hosted on Amazon Web Services (AWS), Atinary’s SDLabs AI platform provides the orchestration layer for a secure, scalable architecture, ensuring seamless machine-to-machine communication at scale. It combines cloud-native compute and secure data environments with modern generative AI (such as Amazon Bedrock) to give bench scientists an intuitive conversational interface to the system.
By focusing initially on small-molecule synthesis and catalysis, specifically complex couplings like the Buchwald-Hartwig and Suzuki reactions, the Atinary Lab in Boston serves as a living blueprint for the future of R&D. It bridges digital simulation and physical reality, running continuous DMTA+L cycles that generate more high-quality, ML-ready data in a single week than a traditional workflow produces in years.
Where This Leads: A Unified Discovery Loop
This is what Atinary’s SDLabs® platform and Self-Driving Lab in Boston are built to deliver: freeing scientists from the mechanics of experimentation to focus on interpretation, analysis, and deciding what to pursue next. The result is a repeatable, sustainable path to the right molecules and performance materials, a competitive advantage for the teams that adopt it, and a faster route to the breakthroughs that pressing global challenges demand, in human health, sustainable food, climate, and clean energy.
Edlyn Wu, PhD (Scientific Solutions Associate)
Mohammed Azzouzi, PhD (Applications Engineer)
Mahrokh Boroujeni (AI Research and Innovation Team Lead)